
Krankheitsbild – Was ist ALS?
Information zur Amyotrophen Lateralsklerose
Was ist Amyotrophe Lateralsklerose?
ALS ist eine Erkrankung des motorischen Nervensystems. Betroffen sind die Nervenzellen, die unsere willkürlichen Bewegungen steuern – das sogenannte erste Motoneuron in der Hirnrinde und das zweite Motoneuron im Rückenmark.
Diese Nervenzellen leiten Signale an die Muskulatur weiter, damit wir uns bewegen, sprechen, schlucken oder atmen
können. Wenn diese Nervenzellen nach und nach ausfallen, erreichen die Signale die Muskeln nicht mehr zuverlässig.
Die Muskeln selbst sind dabei zunächst gesund; sie «verwaisen», weil der Antrieb aus dem Nerv fehlt. Dadurch werden sie schwächer, langsamer oder beginnen zu zucken. Später kommt es zu einem Rückgang der Muskelmasse.
Typisch für ALS ist, dass die Erkrankung immer fokal beginnt – also an einer einzelnen Körperregion, zum Beispiel
an einer Hand, einem Fuss oder in der Sprech- und Schluckmuskulatur.
Erst im Verlauf breitet sie sich weiter aus. ALS betrifft die Motorik, nicht die Sinneswahrnehmungen. Sehen, Hören,
Riechen, Schmecken, Tasten sowie die Funktionen von Herz und Augenmuskulatur bleiben lange stabil.
Fortschreitende Veränderungen – aber sehr individuell
ALS verläuft bei jedem Menschen anders. Die Krankheit ist fortschreitend, doch welche Bereiche zuerst betroffen
sind unterscheidet sich stark von Person zu Person. Viele Betroffene bemerken anfangs nur subtile Veränderungen wie Unsicherheit beim Gehen, eine schnellere Ermüdbarkeit, leichte Schwierigkeiten bei feinmotorischen Bewegungen, Muskelzuckungen oder eine veränderte Sprech- oder Schluckfunktion.
Im weiteren Verlauf kann es zu Einschränkungen beim Gehen, Sprechen, Essen und später auch der Atmung
kommen. In seltenen Fällen können sich auch das Denken oder Verhalten leicht verändern. Nicht alle Funktionen verändern sich im gleichen Ausmass – einige Bereiche bleiben über lange Zeit konstant. Wie schnell oder langsam diese Veränderungen auftreten, lässt sich nicht vorhersagen. Viele Menschen erleben Phasen über Monate oder Jahre, in denen sich die Krankheit nur wenig verändert oder sehr langsam fortschreitet.
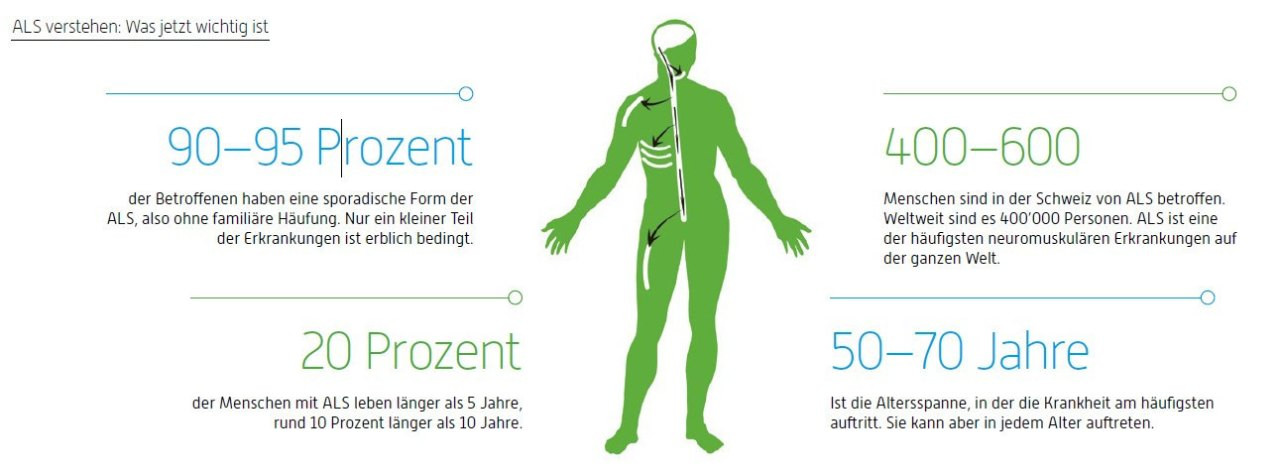
Was ALS im Körper bewirkt
ALS gehört zu den neurodegenerativen Erkrankungen. Das bedeutet, dass bestimmte Nervenzellen im Laufe der Zeit
geschädigt werden. Im Unterschied zu Erkrankungen wie Alzheimer ist bei ALS nur ein klar abgrenzbarer Bereich betroffen – die motorischen Nervenzellen in Gehirn und Rückenmark. Andere Hirnfunktionen bleiben oft lange stabil. Darum bleiben viele ALS-Betroffene trotz körperlicher Einschränkungen geistig fit und in ihrer Wahrnehmung unbeeinträchtigt.
Verlaufsformen
Man unterscheidet zwei Hauptformen des Beginns:
Spinale Form
Beginn in Armen oder Beinen mit Muskelschwäche, Stolpern oder Schwierigkeiten beim Greifen.
Bulbäre Form
Beginn mit Sprech- oder Schluckstörungen, später häufig auch Auswirkungen auf die Extremitäten.
Ursachen: was die Forschung heute weiss
Warum jemand ALS entwickelt, ist wissenschaftlich noch nicht abschliessend geklärt. Die heutige Forschung geht davon aus, dass meist mehrere Faktoren zusammenwirken – darunter genetische Einflüsse, Veränderungen im Zellstoffwechsel sowie mögliche Umweltfaktoren, die noch nicht vollständig verstanden sind. In den allermeisten Fällen tritt ALS ohne familiäre Häufung auf. Niemand trägt Schuld daran, die Krankheit entwickelt zu haben, und auch Lebensstil, berufliche Belastungen oder sportliche Aktivität erklären ALS nicht.

Links