
Caractéristiques de la maladie
Qu’est-ce que la sclérose latérale amyotrophique?
Qu’est-ce que la sclérose latérale amyotrophique ?
La SLA est une maladie du système nerveux moteur. Elle touche les cellules nerveuses qui contrôlent nos mouvements volontaires, à savoir le premier motoneurone dans le cortex cérébral et le deuxième motoneurone dans la moelle épinière. Ces cellules nerveuses transmettent des signaux aux muscles pour nous permettre de bouger, parler, avaler ou respirer. Lorsque ces cellules nerveuses se détériorent progressivement, les signaux ne parviennent plus correctement aux muscles. Au début, les muscles eux-mêmes sont sains, mais ils « s’atrophient » faute de stimulation nerveuse. Cela les affaiblit, les ralentit ou les fait trembler. Plus tard, on observe une diminution de la masse musculaire.
La SLA se manifeste initialement de manière focalisée, c’est-à-dire dans une seule région du corps, par exemple une main, un pied ou les muscles de la parole et de la déglutition. Ce n’est qu’au fur et à mesure de son évolution qu’elle se propage. La SLA affecte la motricité, mais pas les perceptions sensorielles. La vue, l’ouïe, l’odorat, le goût, le toucher ainsi que les fonctions cardiaques et oculaires restent stables pendant longtemps.
Des changements évolutifs, mais très individuels
La SLA évolue différemment chez chaque personne. La maladie est évolutive, mais les zones touchées en premier varient considérablement d’une personne à l’autre. Au début, de nombreuses personnes atteintes ne remarquent que des changements subtils, tels qu’une démarche instable, une fatigue plus rapide, de légères difficultés dans les mouvements de motricité fine, des spasmes musculaires ou des troubles de parole ou de la déglutition.
Au fur et à mesure que la maladie progresse, on peut observer des difficultés à marcher, à parler, à manger et, plus tard, à respirer. Dans de rares cas, la pensée ou le comportement peuvent également subir de légers changements.
Toutes les fonctions ne sont pas affectées de la même manière : certaines restent stables très longtemps. Il est impossible de prédire la rapidité ou la lenteur avec laquelle ces changements vont se produire. De nombreuses personnes connaissent des phases de plusieurs mois ou années pendant lesquelles la maladie évolue peu ou très lentement.
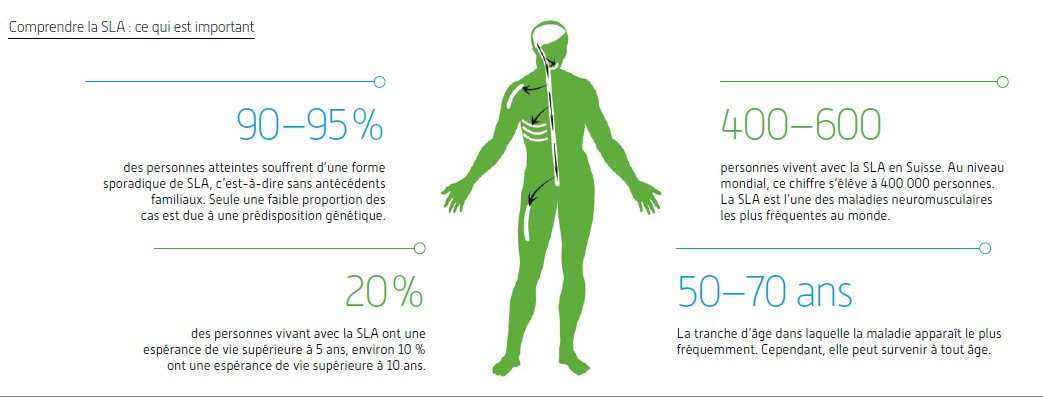
Les effets de la SLA sur le corps
La SLA fait partie des maladies neurodégénératives. Cela signifie que certaines cellules nerveuses sont endommagées au fil du temps. Contrairement à des maladies telles que la maladie d’Alzheimer, la SLA n’affecte qu’une zone clairement délimitée : les cellules nerveuses motrices du cerveau et de la moelle épinière. Les autres fonctions cérébrales restent souvent stables pendant longtemps. C’est pourquoi de nombreuses personnes vivant avec la SLA restent mentalement alertes et ne souffrent d’aucune altération de leurs facultés cognitives en dépit de leurs limitations physiques.
Causes : ce que la recherche permet de savoir aujourd’hui
Les raisons pour lesquelles une personne développe une SLA ne sont pas encore clairement établies scientifiquement. Les recherches actuelles suggèrent que plusieurs facteurs entrent généralement en jeu, notamment des influences génétiques, des modifications du métabolisme cellulaire et d’éventuels facteurs environnementaux qui ne sont pas encore entièrement connus. Dans la grande majorité des cas, la SLA survient sans prédisposition familiale. Personne n’est responsable d’avoir développé la maladie, et le mode de vie, les contraintes professionnelles ou l’activité sportive n’expliquent pas non plus la SLA.
Formes d’évolution
On distingue deux formes principales de survenance de la maladie :
Forme spinale
Apparition dans les bras ou les jambes avec faiblesse musculaire, trébuche-ments ou difficultés à saisir des objets.
Forme bulbaire
Apparition avec des troubles de la parole ou de la déglutition, puis souvent répercussions sur les extrémités.
